
In December 2021, RQM+ acquired AcKnowledge Regulatory Strategies (AcKnowledge RS), a San Diego-based firm specializing in regulatory affairs consulting for the medical device and IVD industry. The integration of this impressive team enhances the extensive RQM+ network of current and former FDA reviewers, scientists, engineers and regulatory and quality experts, and adds additional expertise with FDA submissions. The author of this post is a member of this team, which has done significant work with novel and/or high-risk devices focusing on pre-submissions, 510(k)s, IDEs, PMAs, De Novos, Breakthrough Designation Requests and Safer Technology Program Requests.
The longer you know someone, the more you learn about them. Well, the same goes for medical devices! As knowledge about a medical device increases, we inevitably have a better understanding on its benefits and risks.
As our understanding of a device evolves over time, it is possible that the classification of the device might need a little update. There are two different ways for a medical device to be reclassified at FDA, both of which we review below.
First, there is a way for a medical device to be reclassified as described in the Food, Drug and Cosmetic Act (the Act). Under Section 513(e) of the Act, FDA may reclassify a device based on new information, whether it is discovered on their own, or in response to a petition from an interested person. In this case, “new information” is defined as publicly available, valid scientific evidence. If FDA or a petitioner proposes that a device be reclassified to a lower class (say from the higher risk Class III to a moderate risk Class II), sufficient and valid scientific evidence must be provided in order to support the determination. In July of 2012, the Food and Drug Administration Safety and Innovation Act (FDASIA) was created, which changed the reclassification process to an administrative order process instead of rulemaking. According to FDA’s website that discusses reclassification, in order to reclassify a device under this section of the FD&C Act, FDA must do the following before making the reclassification final:
§ Publish a proposed order in the Federal Register that includes proposed reclassification and summary of the scientific evidence supporting the reclassification
§ Have a panel meeting for the device classification before or after the proposed order has been published
§ Take comments from the public docket into account
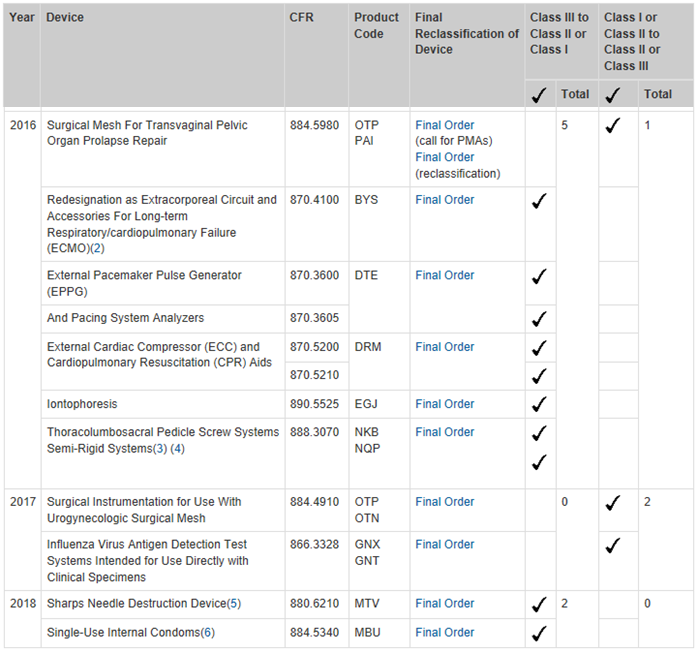
There are also additional rules and guidelines that apply depending on the original class level, and the new class level for a device. For example, for devices being reclassified from Class II to Class III, the scientific evidence must indicate that the general and special controls are insufficient to provide reasonable assurance of safety and effectiveness. Conversely, if a device is being reclassified from Class III to Class II, the scientific evidence must show the opposite; that the general and special controls are sufficient to provide reasonable safety and effectiveness. Lastly, if a device is being reclassified to Class I from either Class III or Class II, the scientific evidence must show that the general controls alone are sufficient to provide reasonable assurance of safety and effectiveness.
The other route for devices reclassification is described in Section 513(f)(3) of the FD&C Act. This originates from the understanding that any device not available before the Medical Device Amendments in 1976 was automatically classified as Class III. This classification was determined regardless of any risk the device posed, and without undergoing any FDA rulemaking processes. These medical devices are more commonly known as postamendment devices. Unlike the process above, these devices may only be considered for reclassification if the initiative comes from FDA or a petition from a manufacturer or importer. Similar to the 513(e) process discussed in the previous paragraph, reclassification of a device can only be made when sufficient regulatory controls can provide reasonable assurance of safety and effectiveness. A device reclassification panel may also be called, should FDA receive a petition requesting reclassification. After all information has been considered, FDA will determine whether the reclassification petition is approved or denied. Following approval, the order describes both the reason for reclassification, as well as any of the device’s risks to patient health.
By the way, if you’re wondering where the whole de novo process fit here, you should check our previous blogs. Keep in mind, a de novo request (per 513(f)(2) of the FD&C Act) is a file where a submitter can request a new classification altogether. Therefore, we don’t really consider it a ‘reclassification’ per the definition of our subject in this blog.
To see a full list of medical devices that have been reclassified since 2013, visit this link. If you’re wondering why the list only goes back five years, it’s because it wasn’t until FDASIA was enforced in 2012 that FDA was required to annually post the devices reclassified in the year prior.
Additional Reading:

.svg "white RQM logo")




